1.2. DNA注释自动化绘图(By 王頔)¶
1.2.1. software installment¶
本软件在dna-features-viewer的基础上进行了二次开发。其依赖的软件包包括:dna_features_viewer、bokeh、pandas和bcbio-gff。建议在anaconda环境基础上进行。 建议参考资料:
- https://pypi.org/project/dna-features-viewer/0.1.8/
- https://github.com/Edinburgh-Genome-Foundry/DnaFeaturesViewer
- https://github.com/VoigtLab/dnaplotlib
作者:王頔 朱振昊
指导教师:夏梦雷
- pip install dna_features_viewer
- pip install bokeh
- pip install pandas
- pip install bcbio-gff
将两个py文件放到当前文件夹或者python搜索路径即可。
1.2.2. 核心功能及用法¶
1.2.2.1. Gff_object¶
本对象可以实现基因位置、类型等信息的快速绘制。 该对象包含9个属性:
- ID,代表序列ID,通常是染色体的ID,每条染色体拥有一个唯一的ID。默认值为:‘chrom1’
- Source,代表基因结构的来源,可以是数据库的名称,比如来自genebank数据库也可以是软件的名称,比如用Genescan软件预测得到,当然,也可以为空,用.点号填充。默认值为:‘custom’
- type,代表区间对应的特征类型,比如gene , exon等。默认值为:‘backbone’
- start,代表区间的起始位置。默认值为:0
- end,代表区间的终止位置。默认值为:1
- score,软件提供了统计值,如果没有,就用.填充。默认值为:‘.’
- strand,代表正负链的信息,+表示正链,-表示负链,?表示不清楚正负链的信息,当正负链信息没有意义时,可以用﹒填充。默认值为:‘+’
- phase,当描述的是CDS区间信息时,需要指定翻译时开始的位置,取值范围包括0,1,2。默认值为:‘.’
- name,默认值为:‘None_name’
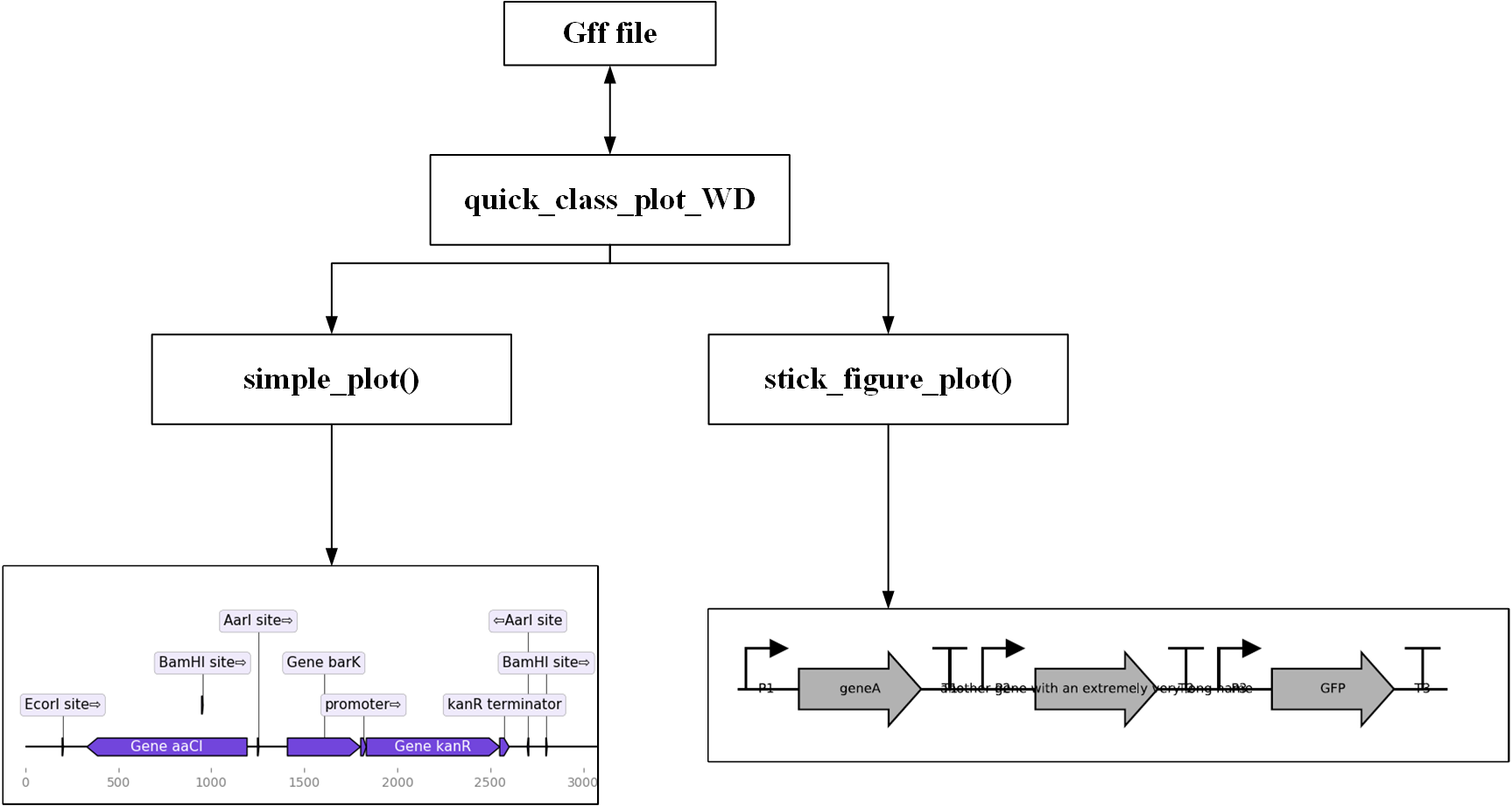
1.2.2.1.1. 核心方法¶
- object_quick_define(a,b,c)
- 用途:用于快速生成一个Gff_Object对象
- 参数: a:int类型,基因的起始位置 b:int类型,基因的终止位置 c:字符串类型,基因的名称
- 输出: 输出一个含有基因起始位置a、基因终止位置b和基因名称c的Gff_Object默认对象
- gff_write(a)
- 用途:用于将生成的Gff_object对象写入Gff文件 参数: a:如果省略,则追加一个新对象;如果为’w’则重新写入
- object_delete(self)
- 用途:根据Gff_object的name 属性将对象进行删除
- txt_generation()
- 用途:方法类属性,将Gff对象信息汇总成gff格式描述方式
- simple_plot()
- 用途:绘制简易图
- stick_figure_plot
- 用途:绘制指示功能模块的简易图
1.2.2.1.2. 简单案例¶
from quick_class_plot_WD import Gff_Object
# 导入quick_class_plot包
xia=Gff_Object()
# 建立一个Gff_Object对象
xia.object_quick_define(200,300,'Gene A')
xia.gff_write('w')
# 绘制一个从200bp到300bp的基因A,并写入gff文件
xia.object_quick_define(400,425,'promoter')
xia.label='promoter'
xia.gff_write()
# 在400bp到425bp绘制一个启动子,并将其写入
xia.object_quick_define(425,500,'GeneB')
xia.strand='-'
xia.gff_write()
# 绘制一个从425bp到500bp的基因B,位于负链,并写入gff文件
Gff_Object.simple_plot()
#绘制具有指示功能模块的简易图
xia.stick_figure_plot(xia.ID, 0, 500)
# 由于stick figure中不含有backbone,如果有backbone,那么就不绘制图案
# 为了美观,可在Gff_object后面加上labelsize=字符串大小,调整标注字体大小
1.2.2.2. gb_features_plot¶
本函数可以基于genebank文件,进行自动绘制。与Gff_object相比,更加的简单。只需要了解7个函数即可(也就是6种绘图方式)
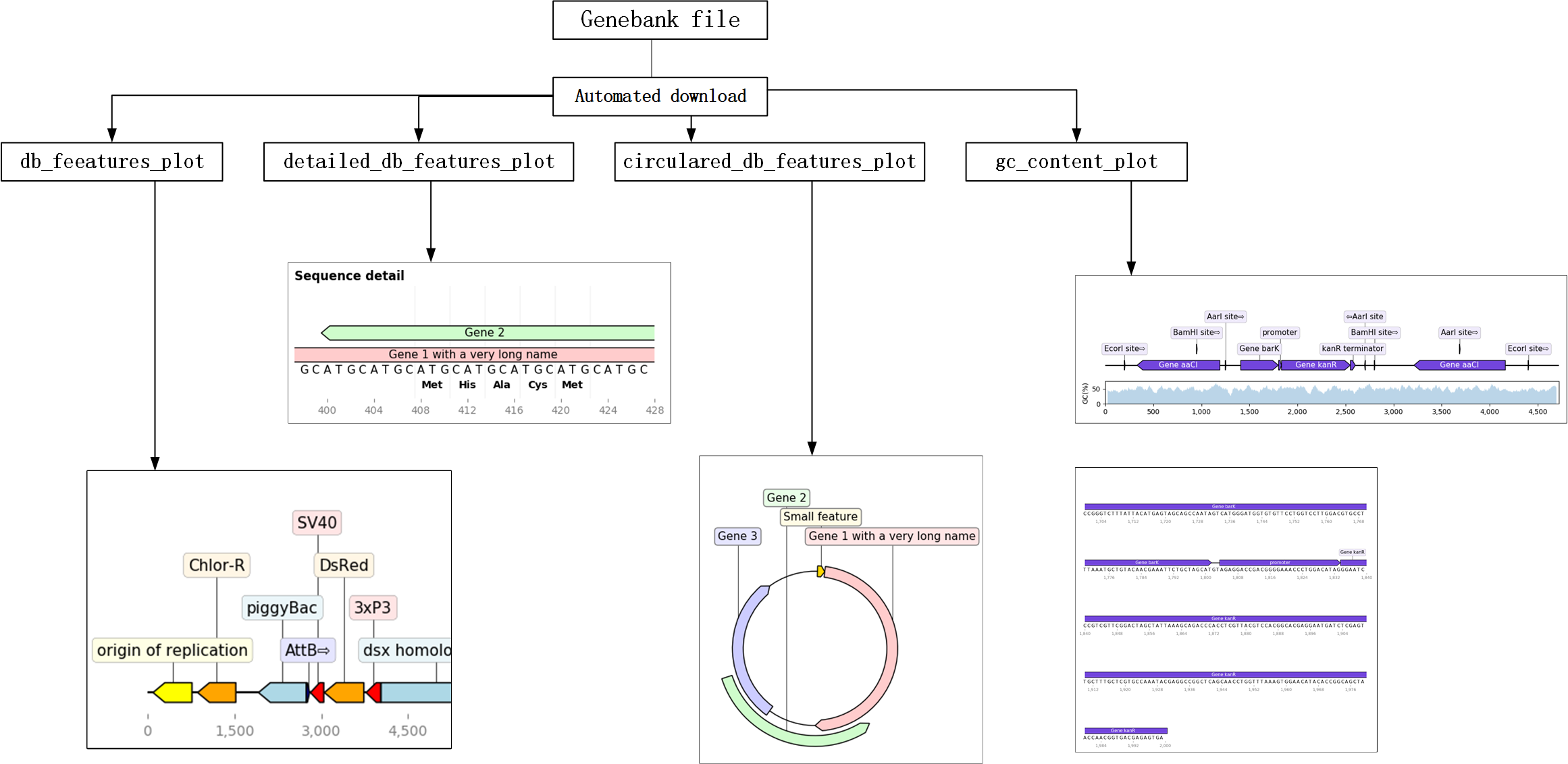
1.2.2.2.1. 核心用法¶
- file = file_get()
- 用途:用于自动下载或打开本地gb文件
- 如果以NCBI的基因号,则自动下载;如果无参数,则通过文件对话框,打开本地文件
- db_feeatures_plot(file)
- 用途:直接绘制gb文件的特征图
- 参数:file为gb文件的路径,类型为字符串
- detailed_db_features_plot(file, a, b)
- 用途:绘制详细的特征图
- 参数:file为gb文件的路径,类型为字符串 a为所关注基因的起始位置,类型为int b为所关注基因的终止位置,类型为int
- circulared_db_features_plot(file)
- 用途:绘制圆形的特征图
- 参数:file为gb文件的路径,类型为字符串
- gc_content_plot(file)
- 用途:绘制包含基因特征和gc含量的图片
- 参数:file为gb文件的路径,类型为字符串
- mult_line_page_plot(file)
- 用途:绘制详细基因信息,并导出pdf格式(也可以直接修改为想要的输出格式)
- 参数:file为gb文件的路径,类型为字符串
1.2.2.2.2. 简单案例¶
from gb_features_plot import *
file = file_get() #通过对话框打开带有注释的gb文件
db_feeatures_plot(file)
detailed_db_features_plot(file, 200, 250)
circulared_db_features_plot(file)
gc_content_plot(file)
mult_line_page_plot(file)
os.startfile("multiline_plot.pdf") # 输出为pdf格式